| |

Gene LibraryAn assortment of several DNA sequences from an organism that has been cloned onto a vector for convenience in purifying, storing, and analyzing make up a gene library. Gene libraries are collections of cloned DNA created with the goal of making it very likely that any specific fragment of the source DNA will be found in the collection. Gene libraries are used to gather and store information as a collection of DNA molecules, much like standard libraries. A specific biological system of interest is represented by a collection of DNA fragments found in all gene libraries. In the 3 billion base pair DNA sequence that makes up the human genome, around 25.000 genes are present. To investigate it, a researcher must first separate each of these genes from the other genes in an organism's DNA. The DNA segments that interest researchers may be located and isolated for further examination. A gene is considered to have been "cloned" when it has been found and replicated. What is Genome?A genome is considered to include all of an organism's genetic material in the sciences of molecular biology and genetics. It is made up of DNA (or RNA in the case of RNA viruses) nucleotide sequences. Protein-coding and non-coding genes, other valuable genomic regions including regulatory sequences (see non-coding DNA), and often a sizable amount of junk DNA with no known function are all found in the nuclear genome. In almost all eukaryotes, the mitochondria and the small mitochondrial genome are present. In addition, algae and plants may include chloroplasts that contain a chloroplast genome. Genomic research is referred to as genomics. Numerous creatures' genomes have had their sequences and annotations completed. Although the original "finished" sequence was missing 8% of the genome, mostly composed of repetitive sequences, the Human Genome Project began in October 1990. It published the sequencing of the human genome in April 2003. Thanks to advancements in technology that could handle sequencing of the many repeated sequences found in human DNA but not fully revealed by the first Human Genome Project research, scientists published the first end-to-end sequence of the human genome in March 2022. HistoryFrederick Sanger, a two-time Nobel Prize laureate, completely sequenced the first DNA-based genome in 1977. Sanger and his research group built a library of the bacteriophage phi X 174 for use in DNA sequencing. Because of the importance of this accomplishment, there is an increasing need for genome sequencing for studies including gene therapy. Now, teams may catalogue variations in genomes and investigate probable genes that may be responsible for disorders including Parkinson's disease, Alzheimer's disease, multiple sclerosis, rheumatoid arthritis, and Type 1 diabetes. These are brought about by the advancement of genome-wide association studies and the accessibility of genomic libraries for the construction and sequencing. Some of the few approaches previously used were linkage studies and candidate gene investigations. Types of Gene LibraryThere are two types of gene libraries:
1) Genomic LibraryA genomic library is a collection of overlapping DNA strands that collectively comprise a single organism's whole genome. Each identical vector in the population holds a separate DNA insert as it stores the DNA. The DNA of the organism is taken out of the cells, digested using a restriction enzyme to separate it into pieces of a certain size, and then assembled into a genomic library. DNA ligase is subsequently used to introduce the pieces into the vector. Then, with just one vector molecule present in each cell, the vector DNA may be assimilated by a host organism, which is often a population of Escherichia coli or yeast. It is simple to amp up and get certain clones from the library for investigation when the vector is carried by a host cell. There are several vector types and insert capabilities to choose from. In general, libraries created from species with bigger genomes need vectors with larger inserts, necessitating the use of fewer vector molecules overall. To determine the appropriate number of clones required for complete genome coverage, researchers may pick a vector while also considering the optimum insert size. Sequencing applications often employ genomic libraries. The human genome and other model species' whole genomes have all been sequenced thanks in large part to their contributions. Construction of Genomic Library It takes a lot of recombinant DNA molecules to build a genomic library. A restriction enzyme is used to digest the genomic DNA of an organism once it has been extracted. The digested pieces for species with extremely tiny genomes (10 kb) may be distinguished by gel electrophoresis. Then, the divided pieces may be isolated and individually cloned into the vector. However, there are too many pieces to remove individually once a restriction enzyme digests a big genome. Separation of clones might take place once the full collection of fragments has been replicated together with the vector. In each situation, the fragments are joined together using the same restriction enzyme-digested vector. The vector may then be used to implant the genomic DNA pieces into a host organism. The procedures for building a genomic library from a big genome are listed below:
Cleaving mechanisms for DNA(a) Physical procedure Using a narrow-gauge syringe needle or sonication, genomic DNA is mechanically sheared into manageable fragment sizes that may be used for cloning. An average DNA fragment size of roughly 20 kb is often preferred for cloning onto-based vectors. DNA fragments may have a range of sizes because DNA fragmentation is unpredictable. A lot of DNA is needed for this approach. (b) The enzyme technique
As detailed below, partial restriction digestion is accomplished by utilizing restriction enzymes that result in blunt or sticky ends: i) Enzymes that cause blunt ends during restriction The genomic DNA may be broken down by restriction enzymes that produce blunt ends, such as HaeIII and AluI. Before cloning, blunt ends are changed into sticky ends. These blunt-ended DNA fragments may be joined to oligonucleotides termed linkers, which include a restriction enzyme recognition sequence, or adaptors, which have an overhanging sticky end for cloning into specific restriction sites. Linkers Linkers are short, double-stranded DNA segments with recognition sites for restriction enzymes that range in length from 8 to 14 bp. By using the ligase enzyme, linkers are joined to blunt-end DNA. In comparison to blunt-end ligation of bigger molecules, linker ligation is more effective. The ligated DNA may be digested with the right restriction enzyme, producing the cohesive ends needed for cloning in a vector. The target DNA segment may include the restriction sites for the enzyme required to produce cohesive ends, which may restrict their usage for cloning. Adapters These are brief oligonucleotide segments with cohesive ends or a linker that restriction enzymes have digested before ligation. The blunt ends of a DNA molecule are changed into cohesive ends by the addition of adaptors. Uses of Adaptors- (a) The actual composition of an adapter, displaying the altered 5'-OH terminus; (b) The transformation of blunt ends into sticky ends by the attachment of adaptors. Following the insertion of the adaptors, the anomalous 5?-OH terminus is changed to the natural 5?-P form by the enzyme polynucleotide kinase, resulting in a sticky-ended fragment that can be put into the proper vector. ii) Sticky end-producing restriction enzymes With the aid of readily accessible restriction enzymes that produce sticky ends, genomic DNA may be digested. For instance, cleaving a vector with BamHI (recognition sequence 5'-GGATCC-3') yields DNA fragments compatible with the sticky end generated by the digestion of genomic DNA with the restriction enzyme Sau3AI (recognition sequence 5'-GATC-3'). The DNA fragments are created and then cloned into an appropriate vector. 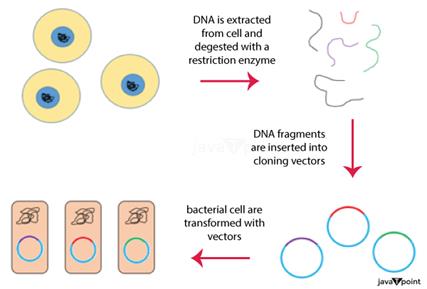
ApplicationsAn organism's genome may be sequenced when a library is established to understand how genes influence an organism or to compare the genomes of related animals. Candidate genes for a variety of functional qualities may be found using the genome-wide association studies discussed above. Genomic libraries may be used to extract genes, which can then be employed on human cell lines or animal models for further research. Additionally, developing high-fidelity clones with precise genome representation and no stability problems would be beneficial as intermediates for shotgun sequencing or the examination of whole genes in functional analysis. 1) Sequential Hierarchy Compared to hierarchical shotgun sequencing, whole genome shotgun sequencing. One of their main applications is using genomic libraries for hierarchical shotgun sequencing, also known as top-down, map-based, or clone-by-clone sequencing. Before high throughput sequencing technology was available, this method was developed in the 1980s for sequencing complete genomes. It is possible to shear individual clones from genomic libraries into smaller segments, typically 500-1000 bp, which are easier to handle for sequencing. After a genomic library clone has been sequenced, the sequence may be used to search the library for other clones with inserts that overlap the sequenced clone. A contig may then be formed by sequencing any additional overlapping clones. It is possible to sequence complete chromosomes using this method, known as chromosome walking. Another way to sequence a genome without using a library of high-capacity vectors is using whole genome shotgun sequencing. Instead, short sequence reads are stitched together using computer algorithms to cover the complete genome. For this reason, genomic libraries and whole genome shotgun sequencing are often employed together. A high-resolution map may be produced by sequencing the inserts from several clones in a genomic library on both ends. The sequences on this map are separated by established distances, making assembling sequence data obtained from shotgun sequencing easier. A BAC library and shotgun sequencing were used to construct the human genome sequence, which was deemed complete in 2003. 1) Genome-wide association research The general use of genome-wide association studies is to identify certain gene targets and polymorphisms within the human population. In order to collect and use this data, scientists, and organizations from many nations partnered to form the International HapMap project. In order to understand similarities and variances within chromosomal areas, this effort compares the genetic sequences of various people. These characteristics are being cataloged by researchers from all of the participating countries using information from people of African, Asian, and European heritage. Such genome-wide analyses help future teams concentrate on coordinating medications with genetic traits in mind while also paving the way for further pharmacological and diagnostic treatments. In genetic engineering, these ideas are already being used. For instance, a research team has really built a PAC shuttle vector that generates a library that represents two-fold coverage of the human genome. This might be a priceless tool for pinpointing the gene or gene set(s) responsible for a certain illness. Furthermore, as shown by the research on baculoviruses, these investigations may be a potent means of examining transcriptional regulation. Overall, improvements in DNA sequencing and genome library assembly have made it possible to efficiently identify a variety of molecular targets. The use of new medication candidates may be accelerated by assimilating these properties using such effective ways. Advantages of Genomic Library
2) cDNA LibraryA "library" of cloned complementary DNA (cDNA) fragments that have been introduced into a group of host cells to make up a section of the organism's transcriptome is known as a "cDNA library." Only the expressed genes of an organism are included in cDNA, which is created from completely transcribed mRNA located in the nucleus. Tissue-specific cDNA libraries may be created in a similar manner. Since the mature mRNA is already spliced in eukaryotic cells, the resulting cDNA is intron-free and may be easily expressed in a bacterial cell. Despite the fact that gene products may readily be recognized, enhancers, introns, and other regulatory elements contained in a genomic DNA library are not included in the information in cDNA libraries, making them less effective and helpful as a tool. cDNA Library Principle
cDNA Library Construction Reverse transcriptase is used to convert mature mRNA from a eukaryotic cell into cDNA. An extended sequence of adenine nucleotides known as the poly-(A) tail, which differentiates mRNA from tRNA and rRNA in eukaryotes, may be utilized as a primer site for reverse transcription. The issue with this is that certain transcripts, like those for the histone, do not encode a poly-A tail. Extraction of mRNA The first step in making cDNA libraries is to extract the mRNA template. The integrity of the separated mRNA should be taken into account since mRNA only includes exons, ensuring that the protein encoded can still be generated. The size of isolated mRNA should be between 500 and 8 kb. Several techniques, including triazole extraction and column purification, are available for RNA purification. mRNA characteristics, such as having a poly-A tail, may be taken advantage of in column purification by utilizing oligomeric dT nucleotide-coated resins, where only mRNA sequences bearing this property will bind. After being bound to the column, the required mRNA is eluted. Construction of cDNA An oligo-dT primer, which is a short strand of deoxy-thymidine nucleotides, is attached to the poly-A tail of the RNA after the mRNA has been purified. The reverse transcriptase enzyme needs the primer to start synthesizing DNA. As a consequence, RNA-DNA hybrids are produced in which a single complementary DNA strand is coupled to an mRNA strand. The RNAse H enzyme is utilized to remove the mRNA by cleaving its backbone and producing free 3'-OH groups, which are crucial for the replacement of mRNA with DNA. The cleaved RNA then serves as a primer for DNA polymerase I, allowing it to recognize and begin replacing RNA nucleotides with DNA. Next, DNA polymerase I is introduced. This is given by the sscDNA itself, which creates a hairpin loop at the 3' end by coiling on itself. The polymerase lengthens the 3'-OH end, and the S1 nuclease's scissor action later opens the loop at the 3' end. The sequences are subsequently cloned onto bacterial plasmids using restriction endonucleases and DNA ligase. The chosen microorganisms are then usually chosen via antibiotic selection. After being chosen, stocks of the bacteria are made, allowing for further growth and sequencing to build the cDNA library. cDNA Library Uses Since cDNA libraries include a significant proportion of non-coding regions, they are often employed to reproduce eukaryotic genomes since they lower the quantity of information present. Expression of eukaryotic genes in prokaryotes is accomplished using cDNA libraries. Introns are not found in the DNA of prokaryotes. Hence they lack the enzymes necessary to remove them during transcription. Since cDNA lacks introns, prokaryotic cells are able to express it. Whereas extra genomic information is less beneficial in reverse genetics, cDNA libraries are most useful in this field. The identification of genes based on the function of the encoded protein is another common application of cDNA libraries in functional cloning. Complementary DNA (cDNA) is used to build expression libraries when analyzing eukaryotic DNA, which helps to confirm that the insert is indeed a gene. cDNA Cloninga. Linkers
b. Restrictions Sites Inclusion
c. cDNA Homopolymer Tailing
How to Screen cDNA Library?
In order to ensure that the probe can attach to any clone with the same sequence, the hybridization procedure is conducted at a temperature that is not stressful. Additionally, non-specific hybridization could happen because some clones resemble that probe only weakly. The probe is washed at a temperature high enough to remove any non-specific clones that have been linked to it. It is vital to make sure that the temperature isn't high enough to clean the probe of clones with sequences that are identical to or equivalent to the probe. As a result, how high of a washing procedure is used depends on whether the probe's origin is homologous or heterologous. Advantages of cDNA Library
Disadvantages of cDNA Library
Genomic DNA Library vs. cDNA Library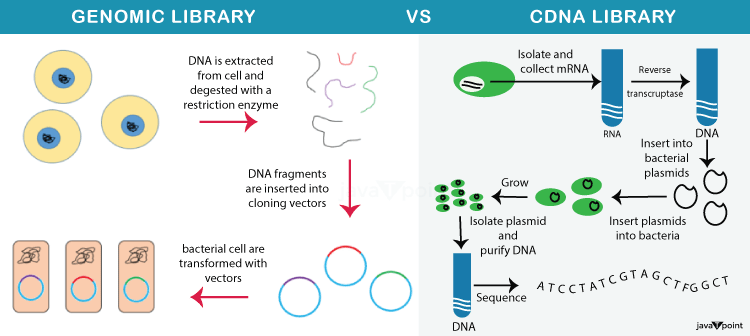
The regulatory and non-coding components of genomic DNA are absent from cDNA libraries. Although they require more resources to create and maintain, genomic DNA libraries provide more specific information about the organism.
Next TopicGene Regulation in Prokaryotes
|
 For Videos Join Our Youtube Channel: Join Now
For Videos Join Our Youtube Channel: Join Now
Feedback
- Send your Feedback to [email protected]
Help Others, Please Share








