| |

Huntington's DiseaseThe neurodegenerative disorder Huntington's disease (HD), commonly known as Huntington's chorea, is mostly hereditary. Mild issues with mood or mental/psychiatric abilities are often the first indications. The result is often a general lack of coordination and a shaky walk. Additionally, chorea, a condition of hyperkinetic movement, is brought on by the illness of the basal ganglia. Uncoordinated, uncontrollable bodily movements associated with chorea become increasingly noticeable as the illness progresses. Physical skills eventually deteriorate so that coordination is difficult to maintain and speech is lost. Mental capacities generally deteriorate into dementia, melancholy, apathy, and impulsivity. The precise symptoms differ a little bit from person to person. The onset of symptoms may occur at any age, although they often appear between the ages of 30 and 50. Each subsequent generation might see the condition progress more quickly. Juvenile HD, which accounts for around 8% of cases that begin before age 20, often manifests as sluggish movement symptoms of Parkinson's disease rather than chorea. Usually, HD is passed down from an afflicted parent with a huntingtin gene (HTT) mutation. However, a novel mutation may cause up to 10% of instances. The huntingtin gene provides the genetic code for the huntingtin protein (Htt). An aberrant mutant protein (mHtt) is produced when the CAG repeats of cytosine-adenine-guanine (sometimes referred to as a trinucleotide repeat expansion) in the gene that codes for the huntingtin protein expand. This protein eventually destroys brain cells in a variety of potential ways. Genetic testing may diagnose a condition at any time, whether or not symptoms are evident. The age at which a person is deemed mature enough to select testing, whether parents have the right to have their children tested, and how to handle confidentiality and disclosure of test results are just a few of the ethical issues this fact brings. 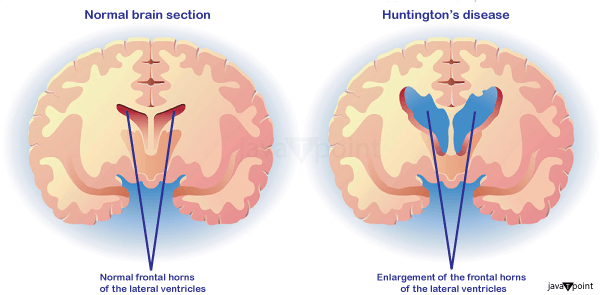
The final stages of HD need full-time care, and there is currently no recognized treatment. Some treatments may reduce symptoms, while others can enhance quality of life. Tetrabenazine has the greatest track record for treating mobility issues. About 4 to 15 persons of European ancestry are affected by HD. It is uncommon among Japanese people, but it is unknown how common it is in Africa. The illness equally impacts both men and women. Although lethal aspiration pneumonia is often mentioned as the condition's final cause of death, complications including pneumonia, heart disease, and physical injuries from falls shorten life expectancy. In around 9% of deaths, suicide is the reason. Death usually happens 15 to 20 years after the illness was first discovered. American doctor Charles Oscar Waters' 1841 description of the illness is considered the first ever. In further depth, the ailment was described in 1872 by American doctor George Huntington. A multinational collaborative effort headed by the Hereditary Disease Foundation uncovered the genetic underpinnings in 1993. In order to encourage research, help people and their families, and raise public awareness, research and support organizations started to emerge in the late 1960s. The exact cause of the disease must be identified, and animal models must be improved; treatments for symptoms or to slow the progression of the disease must be tested, and procedures like stem-cell therapy that aim to replace lost or damaged neurons must be studied. Signs and SymptomsHuntington's disease manifests as a triad of motor, cognitive, and mental symptoms, which most often appear between the ages of 30 and 50 but may start at any age. It is referred to as juvenile Huntington's disease when it manifests early. 50% of the time, psychological symptoms come on first. Early, middle, and late stages of its evolution are often mentioned, along with an earlier prodromal phase. Early personality changes, cognitive and motor abilities difficulties, impatience, and mood swings may all go unrecognized and frequently come before the motor signs. Nearly everyone with HD ultimately displays comparable physical symptoms, although there are considerable individual differences in the onset, course, and severity of cognitive and behavioral symptoms. The most distinctive early physical signs are the jerky, unpredictable, and uncontrolled movements known as chorea. Many people's instinctive motions go unnoticed or are obstructed by them. Chorea may first show up as overall restlessness, involuntary, brief movements that are started but not finished, a lack of coordination, or slower saccadic eye movements. These subtle motor defects typically appear at least three years before more glaring motor dysfunction symptoms. As the disease worsens, the symptoms become more obvious, including stiffness, twisting movements, and aberrant posture. These are indications that there is a problem with the brain system in charge of movement. The progressive impairment of psychomotor processes impacts any movement that needs muscular control. Dystonia is when muscular control is impacted, such as stiffness or muscle contracture. A neurological hyperkinetic movement disorder called dystonia causes twisting or repetitive movements that can resemble tremors. Common side effects include physical trembling, unusual facial expressions, and trouble speaking, eating, and swallowing. Additional symptoms include loss of weight and sleep difficulties. Malnutrition might result from eating problems, which often induce weight loss. In patients with Huntington's disease, weight loss is typical and worsens over time. The Westphal variation of slowness of movement, stiffness, tremors, and seizures is more prevalent in juvenile HD. Juvenile HD often advances at a quicker pace with higher cognitive impairment. 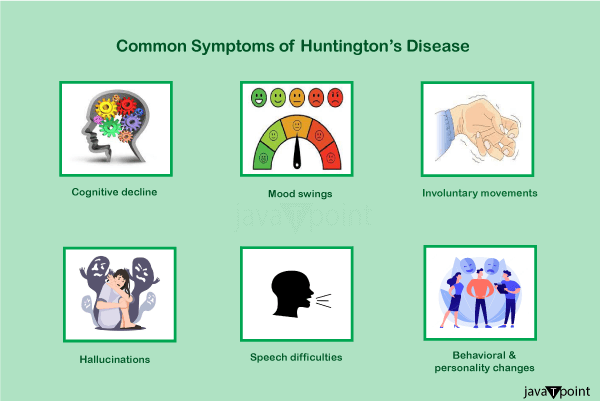
Cognitive abilities gradually deteriorate and eventually turn into dementia. Planning, cognitive flexibility, abstract thought, rule learning, the initiation of suitable activities, and the inhibition of improper acts are executive processes that are particularly impacted. A variety of cognitive impairments may make it difficult to concentrate on activities, lack flexibility, be impulsive, be aware of one's behaviors and skills, and have trouble learning or processing new information. Memory problems often start to show themselves as the illness worsens. The reported memory problems vary from short-term to long-term, including deficiencies in working memory, procedural memory, and episodic memory (remember of one's life). According to reports, neuropsychiatric symptoms include anxiety, despair, a lack of emotional expression, egocentrism, hostility, and obsessive behavior, which may lead to or exacerbate addictions, including drinking, gambling, and hypersexuality. Bipolar disorder, mania, insomnia, and obsessive-compulsive disorder are some more frequent mental conditions. It has also been noted that some individuals have trouble identifying others' unpleasant facial expressions. Studies have shown a wide range in the incidence of these symptoms, with lifetime prevalence rates for mental diseases estimated to range between 33 and 76%. These symptoms are among the most upsetting parts of the illness for many people who have it and their families, often interfering with everyday life and being a factor in institutionalization. A higher risk of suicide results from early behavioral alterations in HD. People often have a diminished awareness of their chorea, cognitive, and emotional deficits. Outside of the brain, mutant huntingtin is distributed throughout the body, and its expression is directly responsible for abnormalities in peripheral organs. These anomalies include testicular atrophy, weight loss, osteoporosis, reduced glucose tolerance, heart failure, muscular atrophy, and testicular failure. Genetics of the Huntington's DiseaseThe huntingtin gene (HTT), which produces the huntingtin protein (Htt), is present in everyone in two copies. The HD gene and the IT15 gene (interesting transcript 15) are other names for HTT. Trinucleotide repeat expansions, small repeats that vary in length across people and might alter length over generations, make up a portion of this gene. A dynamic mutation may increase the number of repeats if the repeat is present in a healthy gene, leading to a faulty gene. The protein known as mutant huntingtin protein (mHtt) is created when the length of this repeating portion exceeds a certain threshold. The different ways these proteins perform lead to pathological alterations, resulting in the illness's symptoms. The condition is caused by a mutation in either person's HTT alleles and is genetically dominant and almost completely penetrant. The length of the repeating part of the gene determines inheritance, not sex. Therefore, the sex of the afflicted parent might impact the severity of the condition. Genetic MutationHD is one of a number of trinucleotide repeat illnesses brought on by repeated sections of a gene that are longer than they should be. At 4p16.3, on chromosome 4's short arm, is where the HTT gene is found. HTT has a trinucleotide repeat, grouping the three DNA bases cytosine, adenine, and guanine (CAG) at various points. A sequence of the three-letter genetic code (codon) CAG, which stands for the amino acid glutamine, produces a chain of glutamine known as a polyglutamine tract (or polyQ tract), as well as the repeated section of the gene known as the polyQ region. People typically have less than 36 repeating glutamines in the polyQ region, which causes the cytoplasmic protein Huntington to be produced. However, creating a protein with distinct properties is triggered by a sequence of 36 or more glutamines. Certain neurons degrade more quickly under the influence of this altered version, known as mutant huntingtin (mHtt). Different parts of the brain depend on various kinds of neurons in different numbers, and as a result, they are impacted differently. About 60% of the difference in the age of symptom onset is explained by the amount of CAG repeats, which is often connected to how much this process is altered. The remainder of the variance results from environmental factors and other genes that affect HD's underlying process. With a considerably later beginning and a more gradual development of symptoms, a reduced-penetrance type of the illness is produced after 36 to 39 repetitions. Sometimes, the onset is so late that the symptoms go unnoticed. Juvenile HD is a kind of HD that may manifest before the age of 20 with very high repetition counts (more than 60). The Westphal variety of juvenile HD is often characterized by stiffness, tremors, and slowness of movement. About 7% of HD carriers are made up of this. InheritanceHuntington's disease is inherited in an autosomal dominant manner, which means that an afflicted person normally receives one copy of the mutant allele-a gene with an enlarged trinucleotide repeat-from an affected parent. Those with a mutant copy of the gene will get the illness due to the high penetrance of the mutation. Each afflicted person's child has a 50% chance of receiving the mutant allele and developing the condition under this inheritance pattern. This probability is gender-neutral. Traits on the X or Y chromosomes are known as sex-dependent or sex-linked genes. Replication instability for trinucleotide CAG repeats with a count of more than 28 becomes worse with each additional repetition. In contrast to replicating a trinucleotide repeat exactly, this often results in additional expansions as generations go by (dynamic mutations). A copy of the gene with an increase in the number of repeats that causes fully penetrant HD may be passed on by an unaffected parent who has an "intermediate" number of repeats (28-35) or "reduced penetrance" (36-40). This is what causes the number of repeats to alter across consecutive generations. Genetic anticipation is the term for the earlier age of illness start and increased severity of disease in subsequent generations caused by increased repetitions. The level of instability is higher during spermatogenesis than during oogenesis; paternally inherited alleles have a larger likelihood of lengthening than maternally inherited ones. Rarely is a novel mutation, in which neither parent has more than 36 CAG repeats, the cause of Huntington's disease. The chance rises to 75% when each parent has two enlarged copies of the HD gene, and to 100% when both parents have expanded copies of the HD gene (which is very unusual). Rare people have both afflicted genes. HD was formerly believed to be the sole illness in which having a second mutant gene did not alter symptoms or progression, but it has since been shown that this is not always the case and may affect both the phenotypic and the pace of advancement. MechanismThe huntingtin protein has a variety of activities and interacts with over 100 different proteins. Although the altered protein's (mHtt) behavior is not fully understood, it is hazardous to several cell types, especially brain cells. As the illness advances, other brain parts, such as the cerebral cortex, are also damaged. The subcortical basal ganglia, first in the striatum, show the most early damage. Early symptoms, such as control over movement, emotion, and higher cognitive function, are caused by the striatum and its links to the cortex's activities. Additionally, HD has altered DNA methylation. Huntingtin FunctionAll cells express Htt, although the brain and testes have the greatest levels, whereas the liver, heart, and lungs only express it to a lesser extent. Although its exact roles are unknown, it does interact with proteins involved in intracellular transport, cell signaling, and transcription. Htt serves a number of purposes in HD-exhibiting genetically altered animals. Since its absence is linked to embryonic mortality in these species, Htt is crucial for embryonic development. The mutant gene is considered to activate caspase, an enzyme involved in the catalysis of apoptosis, by disrupting the ubiquitin-protease system. Additionally, it controls the synthesis of brain-derived neurotrophic factors. This protein safeguards neurons, governs their growth during neurogenesis, and functions as an antiapoptotic agent to stop programmed cell death. Htt also regulates neuronal gene transcription and helps with synaptic vesicular transport and synaptic transmission. When the expression of Htt is lowered, the ensuing features are more like those seen when mHtt is present, but when the expression of Htt is raised, brain cell survival is enhanced, and the effects of mHtt are reduced. Accordingly, it is believed that the sickness is not brought on by insufficient Htt production but rather by a toxic gain-of-function of mHtt in the body. Cellular ChangesThe toxic action of mHtt may produce HD pathology via various cellular alterations. The protein is more likely to cleave into shorter pieces with the polyglutamine expansion in its mutant (polyglutamine-expanded) version. These protein fragments are prone to misfolding and aggregation. As a result, fibrillar aggregates are produced, composed of non-native polyglutamine strands joined by hydrogen bonds from various proteins. These aggregates and other protein deposition disorders share similar cross-beta amyloid geometry. Over time, the aggregates build up within cells to create inclusion bodies, eventually disrupting neural function. Both the cell nucleus and cytoplasm have been discovered to contain inclusion bodies. One of the first pathological alterations is the formation of inclusion bodies in brain cells. While some studies have revealed that inclusion bodies may be poisonous to the cell, others have suggested that they may arise as a protective defense mechanism for the cells. It has been shown that mHtt may kill cells via a number of different routes. These include effects on chaperone proteins, which aid in folding proteins and remove those that are misfolded, interactions with caspases, which are involved in the removal of cells, the toxic effects of glutamine on nerve cells, a reduction in the ability of cells to produce energy and effects on gene expression. It has been discovered that mutant huntingtin protein is crucial for mitochondrial dysfunction. Reactive oxygen species may be released at greater oxidative stress due to mitochondrial electron transport dysfunction. Large concentrations of glutamine are known to be excitotoxic and may harm various cellular structures. HD doesn't have an excess glutamine level, but the altered huntingtin protein interacts with many other proteins in neurons, increasing the susceptibility to glutamine. It is believed that the usual amounts of glutamine will have excitotoxic effects on the increased sensitivity. Macroscopic ChangesThe dorsal striatum in the subcortical basal ganglia is initially the portion of the brain that is most damaged, and cortical involvement spreads laterally to all sections of the brain. The substantia nigra and cortical layers 3, 5, and 6 are among the additional basal ganglia impacted. It is also clear that the hippocampus, Purkinje cells in the cerebellum, lateral tuberal nuclei of the hypothalamus, and certain sections of the thalamus are involved. The size of these regions decreases when cells are lost, depending on their structure and the kinds of neurons they include. Interneurons and spiny cells that project to the internal globus pallidus are less impacted, but striatal medium spiny neurons are more susceptible, especially those with projections towards it. Additionally, HD results in an aberrant rise in astrocytes and activation of the immune cells known as microglia in the brain. The regulation of movement and behavior is largely dependent on the basal ganglia. Although hypotheses suggest they are a member of the motor circuit and the cognitive executive system, their exact roles are unknown. Normally, the basal ganglia obstruct several circuits that produce certain motions. The basal ganglia receives a signal from the cerebral cortex that releases the inhibition to start a certain movement. Damage to the basal ganglia may result in unpredictable and uncontrolled release or restoration of inhibitions, leading to uncomfortable motion initiation, inadvertent motion initiation, or motion halting before or beyond its intended conclusion. The chorea, dyskinesia linked with HD, characterized by unpredictable movements, is brought on by the accumulated damage to this region. Dysphagia, or a decreased capacity to create speech and swallow food and liquids, is always experienced by those afflicted by basal ganglia disorders because they cannot stop motions.
Next TopicPrion Disease
|
 For Videos Join Our Youtube Channel: Join Now
For Videos Join Our Youtube Channel: Join Now
Feedback
- Send your Feedback to [email protected]
Help Others, Please Share








