| |

Bioinformatics DefinitionBioinformatics is an interdisciplinary field that creates techniques and software tools for comprehending biological data, especially when the data sets are big and complicated. In order to analyse and interpret the biological data, bioinformatics, a multidisciplinary discipline of research, incorporates biology, chemistry, physics, computer science, information engineering, mathematics, and statistics. In silico biological query assessments using computational and statistical methods have been carried out using bioinformatics. 
The term "bioinformatics" refers to both biological research that incorporate computer programming into their technique and to particular analysis "pipelines," particularly in the context of genomics. Candidate gene identification and single nucleotide polymorphism analysis are frequent applications of bioinformatics (SNPs). Such identification frequently aims to better understand the genetic basis of disease, special adaptations, desirable traits (especially in agricultural species), or differences between populations. Bioinformatics also seeks to comprehend the proteomic principles that govern the structure of nucleic acid and protein sequences in a less formal manner. From a significant volume of raw data, usable findings can be extracted via image and signal processing. It supports the annotation of genomes and their reported mutations in the field of genetics by assisting with genome sequencing. It has an impact on the creation of biological and gene ontologies for the organisation and search of biological data as well as text mining of biological literature. The examination of the regulation and expression of genes and proteins is another function of it. The comparison, analysis, and interpretation of genetic and genomic data, as well as a broader understanding of the evolutionary elements of molecular biology, are made easier with the use of bioinformatics tools. It aids in the analysis and cataloguing of the biological networks and pathways that are a crucial component of systems biology on a more integrated level. It helps in the simulation and modelling of DNA, RNA, proteins, and biomolecular interactions in structural biology. 
Sequences:Since the Human Genome Project's completion, there has been a significant improvement in sequencing speed and cost. Some labs can now sequence over 100,000 billion bases year, and a complete genome may now be sequenced for $1,000 or less. When protein sequences became available as a result of Frederick Sanger's discovery of the sequence of insulin in the early 1950s, computers became crucial in molecular biology. It proved to be impossible to manually compare multiple sequences. Margaret Oakley Dayhoff was a pioneer in the area. She created one of the earliest databases of protein sequences, which were first published as books, and she invented the techniques for sequence alignment and molecular evolution. Elvin A. Kabat was a pioneer in biological sequence analysis who made a significant contribution to bioinformatics in the early 1970s. Between 1980 and 1991, he published extensive volumes of antibody sequences alongside Tai Te Wu. Bacteriophage MS2 and X174's expanded nucleotide sequences were parsed using informational and statistical algorithms in the 1970s when new DNA sequencing methods were used on them. These research provided evidence that well-known characteristics, such as the coding segments and the triplet code, can be discovered by simple statistical analysis, supporting the idea that bioinformatics can provide useful information. Goals: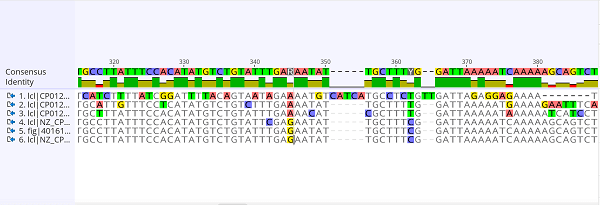
The biological information must be merged to provide a complete picture of these activities in order to examine how common cellular functions are changed in various disease states. The analysis and interpretation of diverse sorts of data is presently the most urgent challenge in the field of bioinformatics as a result of how the subject has developed. Protein domains, protein structures, and nucleotide and amino acid sequences are also included in this. Computational biology is the term used to describe the actual process of data analysis and interpretation. Within bioinformatics and computational biology, significant subfields include:
Increasing our understanding of biological processes is the main objective of bioinformatics. Its emphasis on creating and using computationally complex strategies to accomplish this goal, however, sets it apart from other approaches. Examples include visualisation, data mining, machine learning techniques, and pattern recognition. Sequence alignment, gene discovery, genome assembly, drug design, drug discovery, protein structure prediction, gene expression and protein-protein interaction prediction, genome-wide association studies, modelling of evolution, and cell division/mitosis are some of the major research initiatives in this area. As a result of the management and analysis of biological data, formal and practical difficulties must now be solved through the development of databases, algorithms, computational and statistical techniques, and theory. This is what is referred to as bioinformatics. A significant amount of knowledge on molecular biology has been produced over the past few decades as a result of the rapid advancements in information technology and genomic and other molecular research techniques. These computational and mathematical techniques used to comprehend biological processes are collectively referred to as bioinformatics. In bioinformatics, common tasks include mapping and analysing DNA and protein sequences, aligning DNA and protein sequences for comparison, and building and displaying three-dimensional models of protein structures. With Regard to Other Fields:Although it is frequently mistaken for computational biology, bioinformatics is a branch of research that is related to but different from biological computation. Biological computation builds biological computers using bioengineering and biology, whereas bioinformatics makes use of computation to comprehend biology. Analysis of biological data, especially DNA, RNA, and protein sequences, is a component of both bioinformatics and computational biology. The Human Genome Project and the quick development of DNA sequencing technology were major factors in the discipline of bioinformatics' dramatic expansion that began in the middle of the 1990s. Writing and running computer programmes that employ graph theory, artificial intelligence, soft computing, data mining, image processing, and computer simulation procedures are required for the analysis of biological data to yield relevant information. Theoretical underpinnings including discrete mathematics, control theory, system theory, information theory, and statistics are all used as the basis for the algorithms. Analysis of the Sequence:The DNA sequences of tens of thousands of organisms have been deciphered and preserved in databases since the 1977 sequencing of the Phage -X174. In order to identify genes that encode proteins, RNA genes, regulatory sequences, structural motifs, and repetitive sequences, this sequencing data is processed. A comparison of genes within a species or between various species can reveal relationships between species or similarities in how proteins operate (the use of molecular systematics to construct phylogenetic trees). Analyzing DNA sequences manually has long since been unfeasible due to the expanding volume of data. DNA Sequencing:Sequences must first be collected from the data storage bank, for instance Genbank, before they can be evaluated. Since the raw data may be noisy or impacted by weak signals, DNA sequencing is still a challenging problem. For the many experimental approaches of DNA sequencing, base calling algorithms have been created. Sequence Assembly:The majority of DNA sequencing methods yield small sequence fragments that must be put together to form whole gene or genome sequences. The sequences of countless little DNA fragments are produced by the so-called shotgun sequencing method, which was employed, for instance, by The Institute for Genomic Research (TIGR) to sequence the first bacterial genome, Haemophilus influenzae (ranging from 35 to 900 nucleotides long, depending on the sequencing technology). When a genome assembly programme aligns the ends of these pieces correctly, it can be used to recreate the entire genome because the ends of these fragments overlap. Shotgun sequencing generates sequence data quickly, but for bigger genomes, the process of putting the pieces together might be challenging. Large-memory, multiprocessor computers may need many days to assemble the pieces for a genome the size of the human genome, and the assembled genome typically has many gaps that need to be filled in later. In contrast to chain-termination or chemical degradation methods, shotgun sequencing is the preferred technique for almost all genomes sequenced, and genome assembly algorithms are a crucial area of bioinformatics study. Annotating the Genome:Annotation is the process of labelling the genes and other biological properties in a DNA sequence in the context of genomics. In addition to the requirement to annotate as many genomes as possible because the rate of sequencing is no longer a bottleneck, the majority of genomes are too vast to annotate by hand. As a result, this process must be automated. Although the precise sequence discovered in these sections can differ between genes, the fact that genes have recognisable start and stop areas makes annotation possible. The three levels of genome annotation are nucleotide, protein, and process levels. One of the main functions of nucleotide-level annotation is gene discovery. The most effective approaches for complex genomes combine ab initio gene prediction with sequence comparison with expressed sequence databases and other animals. Genome sequencing can be integrated with other genetic and physical maps of the genome thanks to nucleotide-level annotation. Assigning a function to the genome's byproducts is the main goal of protein-level annotation. Powerful resources for this kind of annotation include databases of protein sequences, functional domains, and motifs. Yet, a new genome sequence typically has half of the predicted proteins with no immediately apparent function. The objective of process-level annotation is to comprehend how genes and their offspring act within the framework of cellular and organismal physiology. The inconsistent terminology employed by various model systems has been one of the challenges in achieving this level of annotation. This issue is being addressed by the Gene Ontology Consortium. The group at The Institute for Genomic Research, which carried out the initial full sequencing and study of the genome of a free-living organism, the bacterium Haemophilus influenzae, published the first description of a comprehensive genome annotation system in 1995. For the purpose of locating the genes that encode all proteins, transfer RNAs, ribosomal RNAs, and other sites as well as for the initial functional assignment, Owen White conceived and created a software system. While most modern genome annotation systems function similarly, there are programmes available for analysing genomic DNA. One such programme is the GeneMark tool, which was developed and used to identify protein-coding genes in Haemophilus influenzae. A new project created by the National Human Genome Research Institute in the U.S. emerged in response to the objectives that the Human Genome Project left unfinished after its termination in 2003. The so-called ENCODE project uses genomic tiling arrays and next-generation DNA-sequencing technologies to collaboratively collect data on the functional components of the human genome. These technologies can automatically produce large amounts of data at a significantly lower cost per base while maintaining the same accuracy (base call error) and fidelity (assembly error). Predicted Gene Function:Despite the fact that sequence homology (and consequently similarity) is the primary criterion used to annotate genomes, it is possible to infer the function of genes using other sequence characteristics. Actually, because protein sequences are more detailed and feature-rich, the majority of approaches for predicting gene function concentrate on them. An example is the prediction of transmembrane regions in proteins based on the distribution of hydrophobic amino acids. But, it is also possible to forecast how a protein will function by using outside data, such as information on protein structure, gene expression, or interactions between proteins. Digital Evolutionary Biology:The study of how different species have changed over time and how they came to be is known as evolutionary biology. By facilitating the following tasks, informatics has aided evolutionary biologists:
The now more intricate tree of life will be recreated in subsequent research. Although the two fields are occasionally mixed up, computational evolutionary biology is not always related to the area of computer science study that employs genetic algorithms. Comparative Genomics:The heart of comparative genome analysis is the determination of the correlation between genes (orthology analysis) or other genomic properties in other organisms. These intergenomic maps allow us to identify the evolutionary events that led to the divergence of two genomes. Genome evolution is shaped by a variety of evolutionary processes operating at different organisational levels. Point mutations have an impact on individual nucleotides at the most fundamental level. Large chromosomal segments go through insertion, deletion, inversion, lateral transfer, duplication, and lateral transfer at a higher level. In the end, processes like endosymbiosis, polyploidization, and hybridization include complete genomes and frequently result in fast speciation. The complexity of genome evolution presents many fascinating challenges for those who create mathematical models and algorithms. These developers turn to a variety of algorithmic, statistical, and mathematical techniques, from exact, heuristic, fixed parameter, and approximation algorithms for problems based on parsimony models to Markov chain Monte Carlo algorithms for Bayesian analysis of problems based on probabilistic models. The assignment of sequences to protein families in many of these investigations is based on the discovery of sequence homology. Global Genomics:Tettelin and Medini first proposed the idea of pan genomics in 2005, and it finally gained traction in the field of bioinformatics. Although initially used to describe closely related strains of a species, pan genome refers to the entire gene repertoire of a specific taxonomic group. It can also refer to a larger context, such as a genus, phylum, etc. The Dispensable/Flexible Genome is made up of genes that are not present in all genomes under research but are present in some or all of them. The Core Genome is the set of genes that are present in all of the genomes under study (these are frequently housekeeping genes essential for survival). The Pan Genome of bacterial species can be characterised using the bioinformatics tool BPGA. Illness Genetics:Infertility, breast cancer, and Alzheimer's disease are just a few of the complex diseases whose genes have been successfully mapped thanks to the development of next-generation sequencing. In order to identify the mutations that cause such complicated disorders, genome-wide association studies are a viable strategy. Through these investigations, tens of thousands of Genetic variations linked to similar diseases and features have been found. Also, one of the most crucial applications of genes is their potential use in prognosis, diagnosis, and treatment. The use of genes to predict the presence or prognosis of disease is the subject of numerous research that explore both the promising methods for selecting the genes to be used as well as the difficulties and hazards involved. Genome-wide association studies have successfully discovered millions of common genetic variations for complex diseases and behaviours; nevertheless, these common variants only account for a small portion of heritability. Some of the missing heritability may be explained by rare mutations. Hundreds of millions of uncommon variations have been found as a result of large-scale whole genome sequencing investigations, which have quickly sequenced millions of complete genomes. The power of genetic association of rare variants analysis of whole genome sequencing studies can be significantly increased by including functional annotations, which help to prioritise rare functional variants and anticipate the effect or function of a genetic variant. With whole-genome sequencing data, some tools have been created that offer all-in-one rare variant association analysis, including integration of genotype data and their functional annotations, association analysis, result summation, and visualisation. In order to find uncommon variations connected to complicated traits, substantial sample sizes must be collected. Meta-analysis of whole genome sequencing research offers an appealing answer to this challenge. Review of Cancer Mutations:Cancer causes complicated, sometimes unforeseen, rearrangements in the genomes of the affected cells. To find previously undiscovered point mutations in a number of cancer-related genes, extensive sequencing efforts are performed. Bioinformaticians continue to develop specialised automated systems to manage the enormous amount of sequence data generated, and they also develop new algorithms and software to compare the sequencing results to the expanding database of human genome sequences and germline polymorphisms. Novel physical detection methods are used, such as single-nucleotide polymorphism arrays for detecting known point mutations and oligonucleotide microarrays for identifying chromosomal gains and losses (a process known as comparative genomic hybridization). When employed at high-throughput to analyse thousands of samples, these detection techniques simultaneously measure hundreds of thousands of sites across the genome, producing terabytes of data for each experiment. Once more, the vast numbers and diverse types of data present bioinformaticians with fresh prospects. In order to infer actual copy number changes, approaches like the Hidden Markov model and change-point analysis are being developed because it is frequently discovered that the data contains a great deal of unpredictability or noise. When analysing cancer genomes bioinformatically for the purpose of finding exome mutations, two key ideas can be applied. Initially, cumulative somatic gene mutations are the cause of cancer. Mutations in the second carcinoma include drivers, which must be differentiated from passengers. Cancer genomics may undergo a significant transformation as a result of the advancements that this next-generation sequencing technology is bringing to the field of bioinformatics. Bioinformaticians can now sequence a large number of cancer genomes swiftly and inexpensively thanks to improved techniques and tools. This could lead to the development of a more adaptable method for analysing cancer-related mutations in the genome and identifying different forms of cancer. In the future, it might be able to follow people as their illness worsens thanks to the sequence of cancer samples. Analysis of lesions discovered to be recurrent among numerous malignancies is another type of data that necessitates fresh informatics development. Expression of Genes and Proteins:Gene Expression Analysis:Numerous methods, such as microarrays, expressed cDNA sequence tag (EST) sequencing, serial analysis of gene expression (SAGE) tag sequencing, massively parallel signature sequencing (MPSS), RNA-Seq, also referred to as "whole transcriptome shotgun sequencing" (WTSS), or various applications of multiplexed in-situ hybridization, can be used to measure the levels of mRNA in order to identify the expression of many genes. Developing statistical tools to distinguish signal from noise in high-throughput gene expression investigations is a prominent research field in computational biology because all of these methods are very noise-prone and/or sensitive to bias in biological measurement. These studies are frequently used to identify the genes associated with a disorder. For example, to identify the transcripts that are up-regulated and down-regulated in a specific population of cancer cells, microarray data from cancerous epithelial cells and data from non-cancerous cells may be compared. Protein Expression Analysis:High throughput (HT) mass spectrometry (MS) and protein microarrays can both give a quick overview of the proteins present in a biological sample. Making sense of protein microarray and HT MS data requires a significant amount of bioinformatics; the former approach faces similar issues as microarrays that target mRNA, while the latter involves the challenge of comparing vast amounts of mass data against predicted masses from protein sequence databases and the challenging statistical analysis of samples where multiple, but incomplete peptides from each protein are detected. Using affinity proteomics, cellular protein localisation in a tissue context can be accomplished. The results are displayed as spatial data based on immunohistochemistry and tissue microarrays. Regulation Analysis:Gene regulation is the intricate orchestration of processes via which a signal, maybe an extracellular signal like a hormone, eventually results in an increase or decrease in the activity of one or more proteins. Bioinformatics methods have been used to investigate different stages of this process. For instance, elements in the genome close by can control how a gene is expressed. The process of promoter analysis entails the discovery and analysis of sequence motifs in the DNA that surrounds a gene's coding region. The degree to which that area is translated into mRNA is affected by these motifs. By interacting in three dimensions, enhancer elements located far from the promoter can also control gene expression. Bioinformatic analysis of chromosomal conformation capture experiments can be used to identify these relationships. Gene regulation can be inferred from expression data by comparing microarray data from several states of an organism and formulating hypotheses about the genes involved in each condition. One could compare different stressors and cell cycle stages in a single-cell organism (heat shock, starvation, etc.). The co-expressed genes can then be identified using clustering methods on the expression data. For instance, it is possible to look for over-represented regulatory elements in the upstream regions (promoters) of co-expressed genes. K-means clustering, self-organizing maps (SOMs), hierarchical clustering, and consensus clustering approaches are a few examples of clustering algorithms used in gene clustering. Structural Bioinformatics: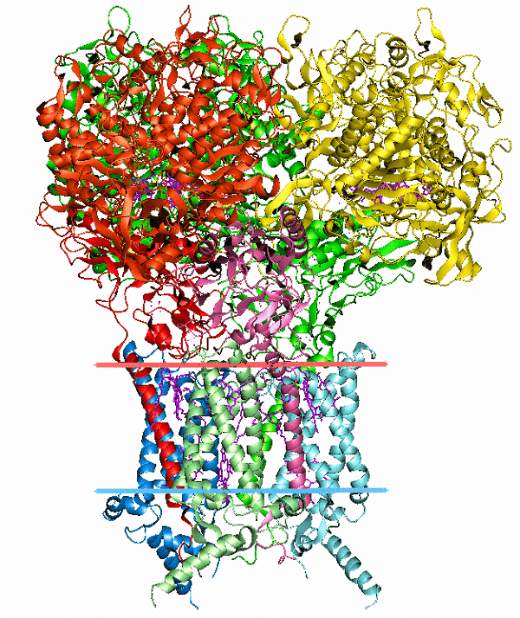
Bioinformatics is used to predict protein structures, which is another crucial application. From the sequence of the gene that codes for it, it is simple to determine the amino acid sequence of a protein, or its so-called fundamental structure. The bulk of the time, a structure in its natural surroundings can be identified only by its fundamental structure. (There are certain exceptions, such the mad cow disease prion, which affects cattle.) To comprehend how the protein works, one must be familiar with its structure. Secondary, tertiary, and quaternary structures are the most common divisions used to classify structural information. Such forecasts still have not found a workable general solution. Heuristics that work most of the time have received the most of the attention thus far. The concept of homology is one of the fundamental concepts in bioinformatics. If the sequence of gene A, whose function is known, is homologous to the sequence of gene B, whose function is unknown, one would assume that B may share A's function. This is how homology is utilised in the genomic branch of bioinformatics to predict the function of a gene. The portions of a protein that are crucial for structure development and interaction with other proteins are identified using homology in the structural branch of bioinformatics. Once the structure of a homologous protein is known, this knowledge is used in a process known as homology modelling to predict the structure of a protein. This has long been the only method for accurately predicting protein structures. Yet with the introduction of new deep-learning algorithm-based software called AlphaFold, created by a bioinformatics team within Google's A.I. research division DeepMind, a paradigm-shifting development occurred. When competing in the 14th Critical Assessment of Protein Structure Prediction (CASP14) computational protein structure prediction software competition, AlphaFold made history by being the first competitor to ever submit predictions with accuracy on par with experimental structures in the vast majority of cases and far outperforming all other prediction software techniques up to that point. The predicted structures for hundreds of millions of proteins have since been made available via AlphaFold. One illustration of this is the haemoglobin found in humans and the leghemoglobin found in legumes, which are distantly related members of the same protein superfamily. Both carry oxygen throughout the body for the same reason. The nearly identical protein architectures of these two proteins, despite the fact that they have entirely distinct amino acid sequences, indicate their nearly identical functions and common progenitor. Protein threading and de novo (from scratch) physics-based modelling are further methods for predicting protein structure. A further application of structural bioinformatics is the use of protein structures for virtual screening models like quantitative structure-activity relationship models and proteochemometric models (PCM). A protein's crystal structure can also be used to simulate various experiments, such as ligand-binding investigations and in silico mutagenesis studies. Biological Networks and Systems:The goal of network analysis is to comprehend the connections that exist within biological networks, such as networks of protein-protein interactions or metabolic pathways. Although a single type of molecule or entity (such as a gene) can be used to build a biological network, network biology frequently aims to integrate numerous data types, including proteins, small molecules, data on gene expression, and others, which are all connected physically, functionally, or both. Systems biology is the study of the interconnectedness of biological processes through the use of computer simulations of the cellular subsystems that make up metabolism, such as the networks of metabolites and enzymes, signal transduction pathways, and gene regulatory networks. With the use of computer simulations of straightforward (artificial) life forms, artificial life or virtual evolution aims to better understand how evolution works. Molecular Interaction Networks: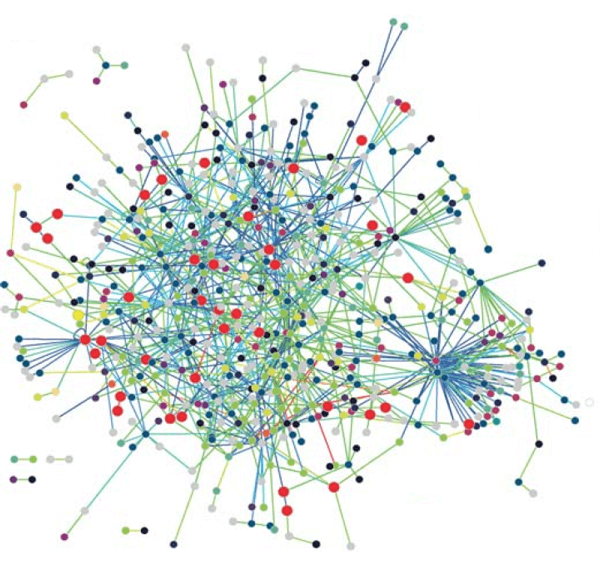
With the help of protein nuclear magnetic resonance spectroscopy (protein NMR) and X-ray crystallography, tens of thousands of three-dimensional protein structures have been identified. A key issue in structural bioinformatics is whether it is practical to predict potential protein-protein interactions solely based on these 3D shapes, without first conducting protein-protein interaction experiments. The protein-protein docking issue has been addressed using a number of different strategies, but it appears that there is still more to be done in this area. Protein-ligand interactions, which may involve drugs, and protein-peptide interactions are further ones that may occur. The core idea behind computer techniques known as docking algorithms for analysing molecular interactions is molecular dynamic simulation of movement of atoms about rotatable bonds. Analysis of the Literature:It has become nearly difficult to read every paper due to the increase in published literature, which has led to fragmented sub-fields of study. The goal of literature analysis is to harvest this expanding corpus of text resources using computational and statistical linguistics. For instance:
Analyzing Images Quickly:Large-scale, high-information-content biomedical picture processing, quantification, and analysis are sped up or completely automated using computational technology. By enhancing accuracy, objectivity, or speed, contemporary image analysis tools help observers make measurements from a large or complicated set of images. The observer might be entirely replaced by a fully developed analytical system. Although these systems are not particular to biomedical imaging, it is becoming more significant for both research and diagnoses. Some examples are:
Databases:For the study and application of bioinformatics, databases are crucial. There are numerous databases that cover different sorts of information, such as DNA and protein sequences, molecular structures, phenotypes, and biodiversity. The most typical combination of the two is for databases to store both empirical data?obtained directly from experiments?and projected data?obtained from analysis. They might be special to a particular organism, process, or interesting molecule. They can also use information gathered from numerous other databases as an alternative. The formats, methods of access, and degree of public accessibility of these databases differ. The following list includes some of the most popular databases. Please go to the link at the start of the subsection for a more complete list.
Software and Tools:There are many different types of software tools for bioinformatics, ranging from straightforward command-line tools to more intricate graphical programmes and stand-alone web services offered by different bioinformatics businesses or government organisations. Totally Free Bioinformatics Applications:Since the 1980s, there have been and continue to be a large number of free and open-source software applications. The potential for creative in silico experiments, the ongoing need for new algorithms for the analysis of evolving classes of biological readouts, and the availability of freely distributable open code bases have all contributed to the creation of opportunities for all research groups, regardless of funding arrangements, to contribute to both bioinformatics and the variety of open-source software available. When used as community-supported plug-ins in for-profit applications, open-source technologies frequently serve as idea incubators. In order to help with the difficulty of bio information integration, they might also offer de facto standards and shared object models. Bioconductor, BioPerl, Biopython, BioJava, BioJS, BioRuby, Bioclipse, EMBOSS,.NET Bio, Orange with its bioinformatics add-on, Apache Taverna, UGENE, and GenoCAD are just a few of the open-source software applications available. Since 2000, the nonprofit Open Bioinformatics Foundation has provided financial assistance for the biennial Bioinformatics Open Source Conference (BOSC) in order to uphold this custom and foster new opportunities. Web-based Bioinformatics services:A wide range of bioinformatics applications have been built with SOAP- and REST-based interfaces, enabling an application operating on a single machine in one region of the world to access methods, information, and computing resources on servers in other regions. Sequence Search Services (SSS), Multiple Sequence Alignment (MSA), and Basic Bioinformatics Services (BSA) are the three categories into which the EBI divides basic bioinformatics services (Biological Sequence Analysis). These service-oriented bioinformatics resources, which range from a collection of standalone tools with a common data format under a single, standalone, or web-based interface to integrative, distributed, and extensible bioinformatics workflow management systems, show the applicability of web-based bioinformatics solutions. Systems for Managing Bioinformatics Workflow:A bioinformatics workflow management system is a particular kind of workflow management system created specially to compose and carry out a workflow, or a set of computational or data manipulation tasks, in a bioinformatics application. The purpose of such systems is to
Bio-Compute and Bio-Compute Objects:Reproducibility in bioinformatics was the topic of a symposium that the US Food and Drug Administration sponsored in 2014 at the Bethesda Campus of the National Institutes of Health. A group of interested parties met frequently over the course of the following three years to discuss the future of the BioCompute paradigm. Representatives of the government, business, and academic sectors were among these stakeholders. A wide range of NIH Institutes and Centers, the FDA, non-profit organizations like the Human Variome Project and the European Federation for Medical Informatics, and research institutions like Stanford, the New York Genome Center, and the George Washington University were all represented by session leaders. It was agreed that the BioCompute paradigm would take the form of digital "lab notebooks" that permit the replication, review, and reuse of bioinformatics techniques. The objective behind this was to promote idea interchange between groups while allowing for more continuity within a research group during periods of typical staff churn. This effort was supported by the US FDA in order to improve the accessibility and transparency of pipeline information for their regulatory staff. In Bethesda, Maryland, at the National Institutes of Health, the group met once more in 2016 to discuss the possibilities for a BioCompute Object, a manifestation of the BioCompute paradigm. Both a preprint manuscript and a "standard trial use" version of this work were uploaded to bioRxiv. Employees, collaborators, and regulators can all access the JSON-sized record thanks to the BioCompute object. Platforms for Education:The computational nature of bioinformatics lends it to computer-aided and online learning in addition to the in-person Masters's degree courses that are taught at many universities. Rosalind and the online courses provided by the Swiss Institute of Bioinformatics Training Portal are two examples of software platforms used to teach bioinformatics principles and techniques. Videos and training session slides are made available on the Canadian Bioinformatics Workshops website under a Creative Commons license. The 4273rd project, often known as the 4273pi project, provides free access to open-source educational resources. The course has been used to instruct both adults and schoolchildren, and it is run on inexpensive Raspberry Pi computers. A group of academics and research personnel working together under the code name 4273 have used Raspberry Pi computers and the 4273 operating system to execute research-level bioinformatics. Online certifications in bioinformatics and related fields are also offered via MOOC platforms, such as the Data Analysis for Life Sciences XSeries on EdX and the Coursera Bioinformatics Specialization (UC San Diego) and Genomic Data Science Specialization (Johns Hopkins) (Harvard). Conclusion:Bioinformatics has developed into a crucial interdisciplinary scientific subject for the biological sciences, supporting "omics" technologies and fields and primarily managing and analyzing "omes" data. The development of high-throughput biological data as a result of "omics" field technological advancements necessitated and prioritized the use of bioinformatics resources, as well as studies and applications for the analysis of complex and further enlarging "Big Data" volumes, which would be impossible and pointless without bioinformatics. As a result, it has been shown here that there is a critical need for the training of highly qualified new-generation scientists who possess integrated knowledge, multilingualism, and cross-field experience and who are able to use sophisticated operating systems, software, algorithms, database/networking technologies, in order to handle, analyze, and interpret high-throughput and rising volumes of complex biological data.
Next TopicBMR Definition
|
 For Videos Join Our Youtube Channel: Join Now
For Videos Join Our Youtube Channel: Join Now
Feedback
- Send your Feedback to [email protected]
Help Others, Please Share








